Introducción a Python y Biopython#
El proyecto de Biopython es una asociación internacional de desarrolladores del lenguaje Python con aplicación a biología molecular computacional. Es de libre acceso y está muy bien documentado, para mayor profundidad consulte el Recetario de Biopython. Para la instalación y requisitos consultar biopython/biopython
Instalación de Python con Conda#
Conda es un gestor de programas que permite descargar de forma ordenada las librerías para un programa y tener múltiples versiones de este. Vamos a usar conda, un paquete que ayuda a gestionar las instalaciones con múltiples ambientes y versiones. Vamos a crear un ambiente (environment) llamado biopython, y allí vamos a instalar las librerías que necesitamos.
conda create --name biopython python=3.9
Ahora activamos el entorno que creamos para instalar los programas
conda activate biopython
conda install -c conda-forge biopython
Ahora podemos “entrar” a python
python
Y chequeamos si Biopython quedó isntalado
import Bio
print(Bio.__version__)
Si sale un error después de llamar Biopython, la instalación fue defectuosa. De lo contrario, está bien por el momento y empezamos a trabajar con secuencias.
Secuencias#
Aunque una secuencia es un conjunto de letras, biopython tiene un formato especial para definir una secuencia
from Bio.Seq import Seq
my_seq = Seq("AGTACACTGGT")
my_seq
my_seq.complement()
my_seq.reverse_complement()
para salir de python escribimos
quit()
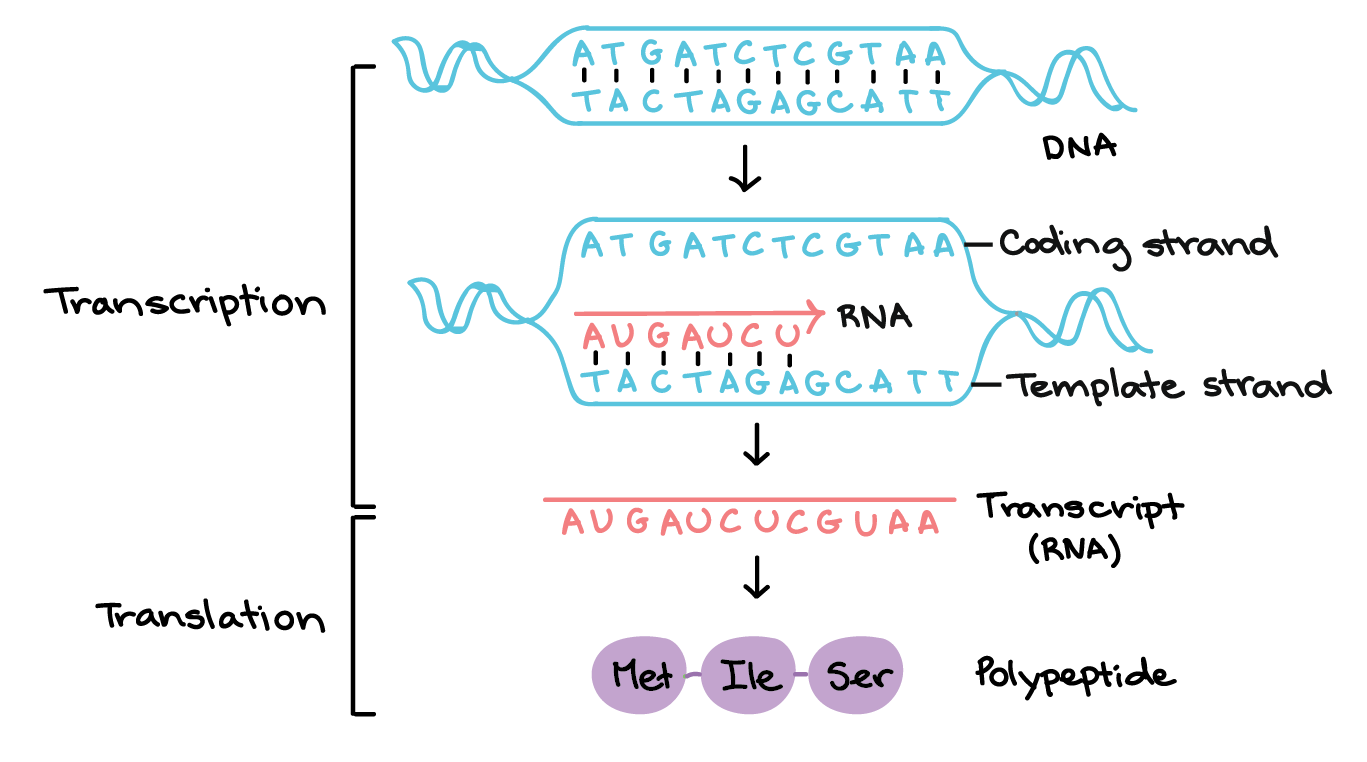
Un recordatorio de la síntesis de proteínas
Formato Fasta de Secuencias#
Existen diferente tipos de formatos para almacenar datos biólogicos, según su naturaleza. Estos formatos son formas estandarizadas que permiten codificar la información de manera simplificada. Muchos de estos formatos son en texto plano, y por lo tanto se pueden visualizar en editores como Notepad++ (Windows) o Sublime Text (Mac). Para el almacenamiento de secuencias se utilizan principalmente dos tipos de formato: FASTA y FASTQ

Vamos a crear un folder para esta clase y a copiar las secuencias descargardas anteriormente de DNA y de proteínas en formatos fasta
mkdir biocomp_python
cp /home/path-to-sequence/sars_covid19/Sars_cov.dna.fa .
cp /home/path-to-sequence/Sars_cov.prot.fa .
El virus COVID-19#
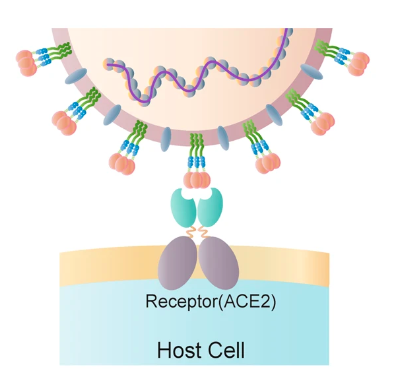
El genoma del virus SARS-CoV-2 está formado por una sola cadena de ARN. Esta cadena condifica para unas pocas proteínas, que son responsables de infectar al hospedero y utilizar su maquinaria enzimática para síntetizar las proteínas del virus y asícumplir su ciclo de vida. Las porteínas virales son generalmente componentes estructurales: envoltura, espícula y membranas. La proteína de la espícula (S) juega un papel fundamnetal en el reconocimiento del receptor y fue crucial para el diseño de las vacunas.
from Bio import SeqIO
dna_file = "Sars_cov.dna.fa"
for record in SeqIO.parse(dna_file, "fasta"):
print(record)
La función SeqIO.parse abre el archivo input_file y lo intepreta como un archivo fasta. Como solo hay una secuencia, el for loop solo itera una vez y muestra (“print”) los atributos de la secuencia. Compárelo con el archivo fasta para entender que significa cada línea.
Ahora vamos a extraer la secuencia y calcular su tamaño
for record in SeqIO.parse(dna_file, "fasta"):
print(record.seq)
print(len(record.seq))
De manera similar se hace con un archivo de múltiples secuencias, por ejemplo el archivo de proteínas. Repita la copia de archivo como se hizo anteriormente, con el de las proteínas. Ahora vamos a iterar sobre las secuencias del archivo “Sars_cov.pep.fa” y a desplegar los identificadores
prot_file = "Sars_cov.prot.fa"
for record in SeqIO.parse(prot_file, "fasta"):
print(record.id)
Cuantas secuencias contiene el archivo? Vamos a imprimir su id, su tamaño y su descripción:
for record in SeqIO.parse(prot_file, "fasta"):
print(record.id,len(record.seq),record.description)