De Genes a Genomas#
Haste el momento hemos partido de genes que conocemos y hemos buscado su secuencia y sus homólogos utilizando bases de datos y alineamientos. Sin embargo, cuando secuenciamos un genoma completo no sabemos de antemano cual es su arquitectura (el orden y ubicación de los elementos del genoma).
“Cracking the code”#
Para extraer la información que contiene un genoma desconocido nos basamos en patrones de secuencias conocidas
Estadísticas de DNA completo#
Algunas características simples de obtener de los genomas son útiles para caracterizarlos:
Longitud#
 By Abizar at English Wikipedia, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=19537795
By Abizar at English Wikipedia, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=19537795Frecuencia de nucleótidos#
La proporción entre A, G, C, and T es una de las propiedades mas fundamentales en la composición de un genoma. En particular el contenido GC
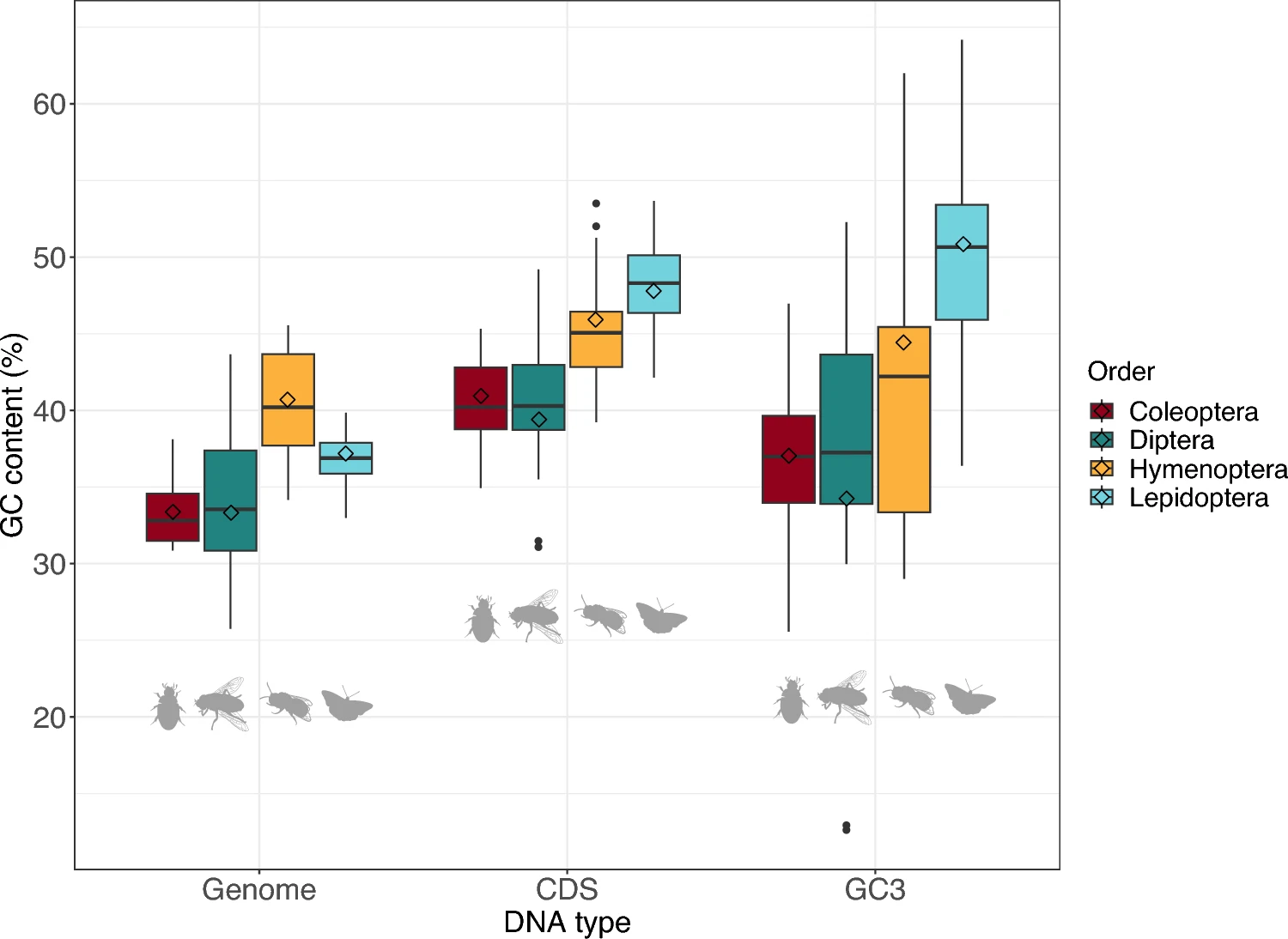 https://doi.org/10.1007/s00239-024-10160-5
https://doi.org/10.1007/s00239-024-10160-5
Ejemplo de estdísticas de genoma en R#
# Mucha parte de este código está basado en
# https://a-little-book-of-r-for-bioinformatics.readthedocs.io/en/latest/src/chapter2.html
# Cargar las librerías
library(seqinr) # si no lo tiene instalado, instale via install.packages("Seqinr")
# Definir el directorio de trabajo
setwd("/Users/laura/docencia/eafit/comp_bio/2024-2/biocomp_genomica/")
# Leer el genoma de covid con función de
covid <- read.fasta(file = "Sars_cov.dna.fa") #archivo descargado previamente y copiado en dir de trabajo
# El primer elemento del objeto es la secuencia
covidseq<-covid[[1]]
# Longitud
length(covidseq)
# Frecuencia de nucleótidos
table(covidseq)
#GC content = (number of Gs + number of Cs)*100/(genome length)
100*(5492+5863)/(8954+5492+5863+9594)
GC(covidseq)
El contenido de GC no solo varía entre especies, sino a lo largo del genoma. Lugares que codifican están caracterizados por tener un mayor contenido de GC (respecto al GC de su genoma) que partes no codificadoras. Para observar la variación local en contenido GC podemos fraccionar el genoma en fragmentos de 5000 nucleótidos, lo cual se denomina “sliding window”
GC(covidseq[1:5000])
GC(covidseq[5001:10000])
GC(covidseq[10001:15000])
GC(covidseq[15001:20000])
GC(covidseq[20001:29903])
En vez de escribir cada función iterativamente podemos incluirla en un for-loop
starts <- seq(1, length(covidseq)-5000, by = 5000) # define una secuencia cada 5000 (ej seq(2,10,by=2))
starts
n <- length(starts) # La longitud de este vector define el número de iteraciones
for (i in 1:n) {
chunk <- covidseq[starts[i]:(starts[i]+4999)] # fragmento de secuencia
chunkGC <- GC(chunk) # contenido GC en fragmento
print (chunkGC)
}
Ahora, en vez de imprimir (print) el contenido GC en cada iteración, lo vamos a almacenar en un vector para graficarlo.
chunkGCs <- numeric(n) # Definimos un vector inicial de 0 con la misma longitud del vector "starts"
# volvemos a iterar en el foor-loop
for (i in 1:n) {
chunk <- covidseq[starts[i]:(starts[i]+4999)] # para un fragmento de nucleótiods de 5000bp "chunk"
chunkGC <- GC(chunk) # hallamos su contenido GC(chunk)
print(chunkGC)
chunkGCs[i] <- chunkGC # y lo almacenamos en el vector chunkGCs reemplazando el 0 por el valor corresponidente a i
}
# vector de starts
starts
# vector de contenido GC
chunkGCs
Ahora vamos a realizar una gráfica que contiene en el eje x a la posición de inicio de la ventana (chunk) de DNA y en el eje j el contenido de GC correspondiente chunkGC
plot(starts,chunkGCs,type="b",xlab="Nucleotide start position",ylab="GC content")
Antes de continuar haremos un Intermezzo sobre cómo escribir una función en R personalizada, para luego aplicarla de nuevo en una gráfica “sliding window” más flexible
# Cómo escribir una función personalizada
# La función se nombra: "mifuncion")
# Se define en terminos de algun(os) parámetro(s) de entrada: x
# Se escribe la salida "return": 20 + x*x
mifuncion <- function(x) { return(20 + (x*x)) } #la definición de la función está entre los corchetes
mifuncion(10)
mifuncion(25)
mifuncion #al escribir una función se muestra su definición
Asimismo podemos definir una gráfica con dos variables (para los ejes x y y) para cualquier tamaño de ventana y cualquier secuencia
slidingwindowplot <- function(windowsize, inputseq) # nombra la función y se definen las variables de entrada
{
starts <- seq(1, length(inputseq)-windowsize, by = windowsize)
n <- length(starts)
chunkGCs <- numeric(n)
for (i in 1:n) {
chunk <- inputseq[starts[i]:(starts[i]+windowsize-1)]
chunkGC <- GC(chunk)
print(chunkGC)
chunkGCs[i] <- chunkGC
}
plot(starts,chunkGCs,type="b",xlab="Nucleotide start position",ylab="GC content")
}
# Llamamos la función
slidingwindowplot(3000, covidseq)
# Podemos guardar esta gráfica en pdf
pdf("covid_sw3000.pdf")
slidingwindowplot(3000, covidseq)
dev.off()